

Producing all-atom protein constructions is a big problem in de novo protein design. Present generative fashions have improved considerably for spine technology however stay tough to resolve for atomic precision as a result of discrete amino acid identities are embedded inside steady placements of the atoms in 3D house. This situation is particularly vital in designing useful proteins, together with enzymes and molecular binders, as even minor inaccuracies on the atomic scale might impede sensible utility. Adopting a novel technique that may successfully deal with these two aspects whereas preserving each precision and computational effectivity is important to surmount this problem.
Present fashions equivalent to RFDiffusion and Chroma focus primarily on spine configurations and supply restricted atomic decision. Extensions equivalent to RFDiffusion-AA and LigandMPNN try and seize atomic-level complexities however are usually not in a position to characterize all-atom configurations exhaustively. Superposition-based strategies like Protpardelle and Pallatom try and strategy atomic constructions however endure from excessive computational prices and challenges in dealing with discrete-continuous interactions. Furthermore, these approaches wrestle with attaining the trade-off between sequence-structure consistency and variety, making them much less helpful for sensible purposes in precise protein design.
Researchers from UC Berkeley and UCSF introduce ProteinZen, a two-stage generative framework that mixes movement matching for spine frames with latent house modeling to attain exact all-atom protein technology. Within the preliminary part, ProteinZen constructs protein spine frames inside the SE(3) house whereas concurrently producing latent representations for every residue by flow-matching methodologies. This underlying abstraction, subsequently avoids direct entanglement between atomic positioning and amino acid identities, making the technology course of extra streamlined. On this subsequent part, a VAE that’s hybrid with MLM interprets the latent representations into atomic-level constructions, predicting sidechain torsion angles, in addition to sequence identities. The incorporation of passthrough losses improves the alignment of the generated constructions with the precise atomic properties, making certain elevated accuracy and consistency. This new framework addresses the restrictions of current approaches by attaining atomic-level accuracy with out sacrificing variety and computational effectivity.
ProteinZen employs SE(3) movement matching for spine body technology and Euclidean movement matching for latent options, minimizing losses for rotation, translation, and latent illustration prediction. A hybrid VAE-MLM autoencoder encodes atomic particulars into latent variables and decodes them right into a sequence and atomic configurations. The mannequin’s structure incorporates Tensor-Area Networks (TFN) for encoding and modified IPMP layers for decoding, making certain SE(3) equivariance and computational effectivity. Coaching is completed on the AFDB512 dataset, which may be very rigorously constructed by combining PDB-Clustered monomers together with representatives from the AlphaFold Database that accommodates proteins with as much as 512 residues. The coaching of this mannequin makes use of a mixture of actual and artificial information to enhance generalization.
ProteinZen achieves a sequence-structure consistency (SSC) of 46%, outperforming current fashions whereas sustaining excessive structural and sequence variety. It balances accuracy with novelty nicely, producing protein constructions which might be various but distinctive with aggressive precision. Efficiency evaluation signifies that ProteinZen works nicely on smaller protein sequences whereas displaying promise to be additional developed for long-range modeling. The synthesized samples vary from a wide range of secondary constructions, with a weak propensity towards alpha-helices. The structural analysis confirms that many of the proteins generated are aligned with the recognized fold areas whereas displaying generalization in direction of novel folds. The outcomes present that ProteinZen can produce extremely correct and various all-atom protein constructions, thus marking a big advance in comparison with current generative approaches.
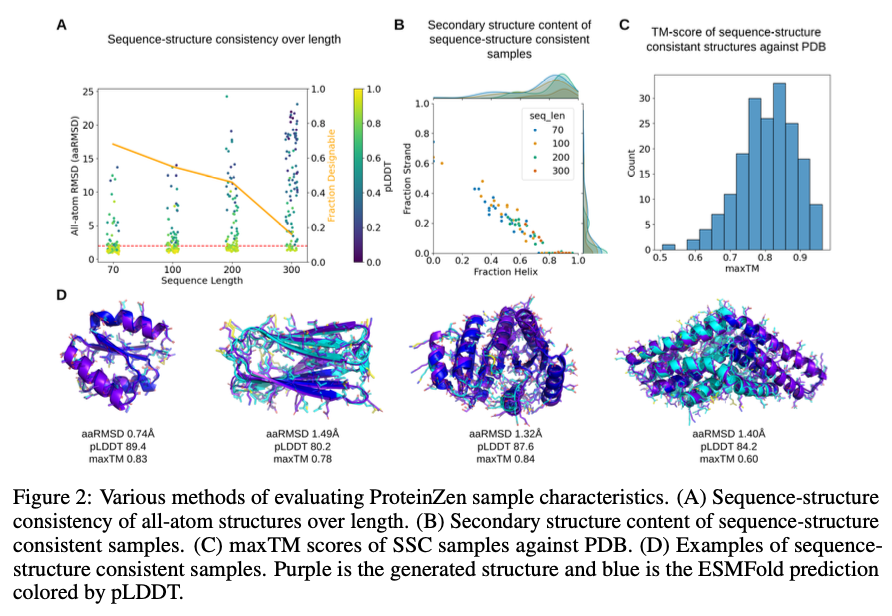
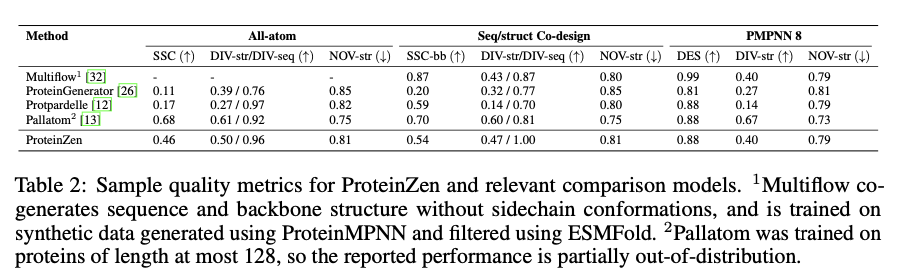
In conclusion, ProteinZen introduces an progressive methodology for the technology of all-atom proteins by integrating SE(3) movement matching for spine synthesis alongside latent movement matching for the reconstruction of atomic constructions. By means of the separation of distinct amino acid identities and the continual positioning of atoms, the approach attains precision on the atomic degree, all of the whereas preserving variety and computational effectivity. With a sequence-structure consistency of 46% and evidenced structural uniqueness, ProteinZen establishes a novel normal for generative protein modeling. Future work will embrace the advance of long-range structural modeling, refinement of the interplay between the latent house and decoder, and the exploration of conditional protein design duties. This growth signifies a big development towards the exact, efficient, and sensible design of all-atom proteins.
Take a look at the Paper. All credit score for this analysis goes to the researchers of this venture. Additionally, don’t overlook to comply with us on Twitter and be a part of our Telegram Channel and LinkedIn Group. Don’t Overlook to hitch our 60k+ ML SubReddit.
🚨 Trending: LG AI Analysis Releases EXAONE 3.5: Three Open-Supply Bilingual Frontier AI-level Fashions Delivering Unmatched Instruction Following and Lengthy Context Understanding for International Management in Generative AI Excellence….
Aswin AK is a consulting intern at MarkTechPost. He’s pursuing his Twin Diploma on the Indian Institute of Know-how, Kharagpur. He’s captivated with information science and machine studying, bringing a robust educational background and hands-on expertise in fixing real-life cross-domain challenges.

